|
|
A library for working with phylogenetic and population genetic data.
v0.32.0 |


|
|
|
A library for working with phylogenetic and population genetic data.
v0.32.0 |
|
Metagenomic studies often need to biologically classify millions of DNA sequences, for example so-called short reads, coming from environments such as water, soil, or the human body. The assignment of those reads to known reference sequences helps to assess the composition and diversity of microbial communities and allows for comparing them.
Phylogenetic (or evolutionary) placement is a method to obtain a classification of metagenomic sequences in terms of a reference phylogenetic tree. The following publications introduce the topic in detail. They are the main programs to obtain phylogenetic placements:
The output of a phylogenetic placement analysis is standardized in the jplace file format [4].
The placement algorithm typically takes as input:

The algorithm finds the most likely (via maximum likelihood) insertion positions for every query sequence on the reference tree. The resulting assignment of a query sequence to a branch is called a placement. A query sequence can have multiple possible placement positions at different branches of the tree, with different likelihoods.
The likelihoods of placement positions are usually transformed into the likelihood weight ratio. For a given query sequence, those values sum up to 1.0 for all branches of the tree. They can thus be seen as a probability distribution of possible placement positions on the tree.
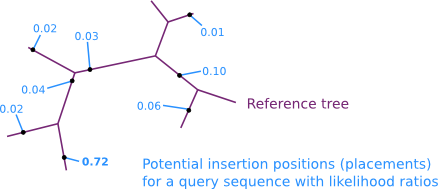
The set of placements for a query sequence is called a Pquery. It contains a name (usually, that is the name of the original query sequence) and the placements with their features (e.g., an ID of the edge where the placement is located, its likelihood, etc.). See [4] for details.
Genesis has classes and functions to work with all relevant data of evolutionary placement. This tutorial focuses on the actual placement data, that is, Pqueries with their placement positions. See the tutorial pages Tree Basics and Sequence for details on those related topics.
The most important class for evolutionary placement is the Sample. A sample is a representation of a whole jplace file: It stores the reference tree and a set of Pqueries.
To read the data from a jplace file into a Sample, use a JplaceReader:
Each Pquery contains the Placements of a query sequence, as well as any Names associated with it.
You can add Pqueries or find them like this:
Removing placements with certain properties (filtering) works like this:
Writing back your results to a new jplace file is done using a JplaceWriter:
See the API reference for details and for more functions and classes related to this topic.
Please refer to the following articles for more information on phylogenetic placement of short reads:
[1]P. Barbera, A. M. Kozlov, L. Czech, B. Morel, D. Darriba, T.Flouri, A. Stamatakis, EPA-ng: Massively Parallel Evolutionary Placement of Genetic Sequences, Systematic Biology, vol. 68, no. 2, pp. 365–369, 2018. DOI: 10.1093/sysbio/syy054
[2]S. Berger, D. Krompass, and A. Stamatakis, Performance, accuracy, and web server for evolutionary placement of short sequence reads under maximum likelihood, Systematic Biology, vol. 60, no. 3, pp. 291–302, 2011. DOI: 10.1093/sysbio/syr010
[3]F. A. Matsen, R. B. Kodner, and E. V. Armbrust, pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree, BMC Bioinformatics, vol. 11, no. 1, p. 538, 2010. DOI: 10.1186/1471-2105-11-538
[4]F. A. Matsen, N. G. Hoffman, A. Gallagher, and A. Stamatakis, A format for phylogenetic placements, PLoS One, vol. 7, no. 2, pp. 1–4, Jan. 2012. DOI: 10.1371/journal.pone.0031009
 1.8.17
1.8.17